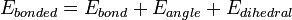Parameterization
In addition to the functional form of the potentials, a force field defines a set of parameters for each type of atom. For example, a force field would include distinct parameters for an oxygen atom in a carbonyl functional group and in a hydroxyl group. The typical parameter set includes values for atomic mass, van der Waals radius, and partial charge for individual atoms, and equilibrium values of bond lengths, bond angles, and dihedral angles for pairs, triplets, and quadruplets of bonded atoms, and values corresponding to the effective spring constant for each potential. Most current force fields use a "fixed-charge" model by which each atom is assigned a single value for the atomic charge that is not affected by the local electrostatic environment; proposed developments in next-generation force fields incorporate models for polarizability, in which a particle's charge is influenced by electrostatic interactions with its neighbors. For example, polarizability can be approximated by the introduction of induced dipoles; it can also be represented by Drude particles, or massless, charge-carrying virtual sites attached by a springlike harmonic potential to each polarizable atom. The introduction of polarizability into force fields in common use has been inhibited by the high computational expense associated with calculating the local electrostatic field.
Although many molecular simulations involve biological macromolecules such as proteins, DNA, and RNA, the parameters for given atom types are generally derived from observations on small organic molecules that are more tractable for experimental studies and quantum calculations. Different force fields can be derived from dissimilar types of experimental data, such as enthalpy of vaporization (OPLS), enthalpy of sublimation (CFF), dipole moments, or various spectroscopic parameters (CFF).
Parameter sets and functional forms are defined by force field developers to be self-consistent. Because the functional forms of the potential terms vary extensively between even closely related force fields (or successive versions of the same force field), the parameters from one force field should never be used in conjunction with the potential from another.


 where the components of the covalent and noncovalent contributions are given by the following summations:
where the components of the covalent and noncovalent contributions are given by the following summations: