Functional form
Further information: Molecular mechanics

Molecular mechanics potential energy function with continuum solvent.
 where the components of the covalent and noncovalent contributions are given by the following summations:
where the components of the covalent and noncovalent contributions are given by the following summations: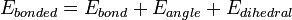

The bond and angle terms are usually modeled as harmonic oscillators in force fields that do not allow bond breaking. A more realistic description of a covalent bond at higher stretching is provided by the more expensive Morse potential. The functional form for the rest of the bonded terms is highly variable. Proper dihedral potentials are usually included. Additionally, "improper torsional" terms may be added to enforce the planarity of aromatic rings and other conjugated systems, and "cross-terms" that describe coupling of different internal variables, such as angles and bond lengths. Some force fields also include explicit terms for hydrogen bonds.
The nonbonded terms are most computationally intensive because they include many more interactions per atom. A popular choice is to limit interactions to pairwise energies. The van der Waals term is usually computed with a Lennard-Jones potential and the electrostatic term with Coulomb's law, although both can be buffered or scaled by a constant factor to account for electronic polarizability and produce better agreement with experimental observations.
No comments:
Post a Comment